Ehlers-Danlos syndrome (EDS) is a group of disorders affecting the connective tissues that support the skin, bones, blood vessels, and many other organs and tissues. The defects that arise in the connective tissue result in the specific symptoms associated with EDS and range from mildly loose joints to life-threatening complications. Initially the different types of EDS were named with roman numerals (EDS I, EDS II, etc.), but in 1997 the Villefrance nosology for the different EDS types was proposed (Beighton et al. 1998), which was revised in 2017 (Malfait et al. 2017). The 2017 International Classification of the Ehlers–Danlos Syndromes has become the generally accepted standard and lists thirteen types, with descriptive names such as classic EDS (cEDS), classic-like EDS (clEDS) and vascular EDS (vEDS), which is also used here.
Vascular EDS is an autosomal dominant disorder solely associated with mutations in the COL3A1 gene. Patients have arterial, intestinal and/or uterine fragility; thin translucent skin that bruises easily; characteristic facial appearance (thin vermilion of the lips, micrognathia, narrow nose, prominent eyes); and an aged appearance to the extremities, particularly the hands. vEDS is a severe disorder: the most prominent signs in patients are vascular dissection or rupture, gastrointestinal perforation, or organ rupture. Approximately 25% of patients experience a severe complication before they reach 20 years of age, and at 40 years ~80% have severe complications. The median age of death is estimated at 50 years. Estimates of the prevalence range from 1:50,000 to 1:200,000. In >95% of patients a COL3A1 mutation can be identified, and >95% of the known COL3A1 mutations are point mutations or small indels that are detected with sequence analysis. 1-2% of the known pathogenic mutations in COL3A1 are large deletions or duplications that can be detected by MLPA (Pepin et al. 2014). More information about vEDS can be found at: https://www.ncbi.nlm.nih.gov/books/NBK1494/ and https://omim.org/entry/130050.
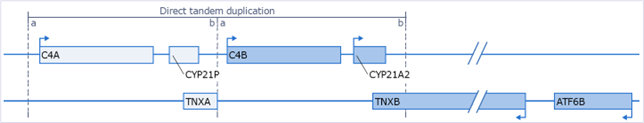
Classic-like EDS is an autosomal recessive disorder associated with mutations in the TNXB gene. clEDS is primarily characterised by hyperextensible skin, generalised joint hypermobility, muscle weakness, intestinal & tracheal tissue weakness, and in some cases mitral valve prolapse, which affects blood flow between the chambers of the heart. clEDS is very rare, but all known patients have mutations in TNXB. Determining the TNXB genotype can be challenging because the gene is located in a complex genomic locus that contains a tandem duplicated region (Fig. 1; based on Gitelman et al. 1992). This cluster of genes (C4, CYP21 and TNX) is also referred to as RCCX module.
TNXB genotyping difficulties are mainly due to the presence of a pseudogene (TNXA), which is almost completely identical to the 3’-end of TNXB (exons 32–44). The only exception is a 120 basepair (bp) TNXB-specific sequence in exon 35. Currently, it is not possible to specifically detect TNXB CNVs in the highly homologous exons 32–34 and 36–44. CNV analysis of exon 35 and of sequences upstream of exon 32 and downstream of exon 44 are used to infer whether deletions or duplications of the highly homologous exons are present. Based on recent research the relative frequency of CNVs among pathogenic TNXB mutations is estimated at ~30%, most of which originate from rearrangements between TNXB and TNXA that include the common 120 bp deletion in exon 35, also described as the c.11435_11524+30 deletion (Demirdas et al. 2017, Green et al. 2020).
Unequal crossovers between the tandem duplicated regions occur frequently and result in chimeric pseudogene-gene products. Bimodular RCCX - as depicted in Figure 1 - has an allele frequency of ~69% and is considered the ‘wild type’ situation, but monomodular and trimodular RCCX structures are also frequent and account for ~17% and ~14%, respectively (Blanchong et al. 2000). In addition, due to the high homology between CYP21A2 and CYP21A1P, but also between TNXB and TNXA, gene conversions are likely to occur in these regions. When the functional gene obtains sequence(s) form the non-functional pseudogene or vice versa this is referred to as a gene conversion. Gene conversions can present as apparent CNVs and can be benign, especially when the pseudogene obtains sequences from the functional gene.
When unequal crossover occurs between CYP21A1P and CYP21A2, it will result in a non-transcribed chimeric gene fusion (CYP21A1P-CYP21A2). When both alleles of CYP21A2 are lost or non-functional this leads to the disease Congenital Adrenal Hyperplasia (CAH). The P155 probemix is not suitable to detect all known CYP21A1P-CYP21A2 gene fusions. To determine the copy number of CYP21A2 and detect more CYP21A1P-CYP21A2 fusion genes the SALSA MLPA Probemix P050 CAH is recommended. An unequal crossover between CYP21A1P and CYP21A2 does not affect the TNXB gene. However, ~10% of CAH patients also have TNXB deficiency. This contiguous gene syndrome is referred to as CAH-X. In all known cases this is the result of unequal crossover between TNXA and TNXB, resulting in a non-functional TNXB-TNXA chimeric fusion gene and deletion of the CYP21A2 gene that is located amidst TNXA and TNXB. The known TNXA-TNXB fusions described in literature (Burch et al. 1997; Morissette et al. 2015) can be detected by the SALSA MLPA Probemix P155 EDS. More information about clEDS can be found at: https://omim.org/entry/606408.